
2x Nobel Prize Winner’s Cure for Heart Disease
Linus Pauling traced the world’s leading killer to a missing vitamin, named the molecule that does the damage, and proposed a cure. Three decades later, cardiology is quietly proving the molecule right.
Dr. Kevin Ham, MD
People are asking me “What about Lipoprotein(a)? Have you heard of this? You have a 20% chance for this genetic factor for atherogenic heart disease.
I find this study on Lp(a) very interesting, even though I’ve gotten it tested twice to see if I could move it. I did, somewhat lower, but it was already normal. I thought that I must have a high Lp(a) which factored in my plaque. I went deep on this one and didn’t edit as much or cut it short because I think it’s something to really think about, even if you have normal Lp(a), you will learn a lot.
2x Nobel Prize Winner
“I think we can get almost complete control of cardiovascular disease.”
Dr. Linus Pauling, 1992
Linus Pauling remains the only person ever to win two unshared Nobel Prizes, Chemistry in 1954 and Peace in 1962. He had mapped the chemical bond and read the structure of proteins. So when, late in life, he turned to the disease that kills more human beings than any other, the scientific world had every reason to listen, and almost none of it did.
Heart disease is the number 1 killer on Earth, the leading cause of death for decades. Pauling made a claim so simple it sounded crazy. Coronary disease, he argued, is at its root a chronic, low grade vitamin C deficiency, and it can be prevented and even reversed with three molecules you can buy at any pharmacy for the price of a coffee.
Consider the scale. Elevated Lp(a), the molecule at the center of his theory, is carried by roughly 1 in 5 people on Earth, more than 1.4 billion human beings, the overwhelming majority of whom have never heard of it or been tested for it. It is one of the most common dangerous traits in our species, hiding in plain sight.
To understand why he believed this, and why his colleagues did not, you have to start two decades before Pauling, in a Canadian hospital, with a doctor almost no one remembers.
The Canadian Who Proved Arterial Plaque Reversal
“The Reversibility of Atherosclerosis.”
Dr. G.C. Willis, Canadian Medical Association Journal, 1957
In the early 1950s a Canadian physician named G.C. Willis fixated on a question the cholesterol theory could not answer. If plaque simply precipitated out of the blood, why did it appear in a precise, repeating pattern, always at the bends and forks of arteries where the pulse hammers hardest? To Willis it looked less like a spill than a repair, the body patching where mechanical stress had cracked the lining.
He had a clue. An earlier investigator, Paterson, had found the tissues of heart patients depleted of vitamin C, and it was textbook that vitamin C is required to build collagen, the protein that holds an artery together. Willis chose his animal with care. The guinea pig, like humans and unlike rabbits, rats, or dogs, cannot make its own vitamin C.
The contrast across the animal kingdom was his strongest clue. Almost every other creature makes its own vitamin C in amounts that dwarf what we eat. A 150 pound goat makes around 13 grams a day, and a feedback system can drive that as much as thirteenfold higher under illness, injury, or hard exertion. Those animals, flooded with their own ascorbate, almost never develop the arterial disease that kills humans. To Willis the lesson was plain, that the wall, kept strong by vitamin C, was what mattered, not the cholesterol passing through it. The few species that lost the enzyme, humans, guinea pigs, and a handful of others, are the ones whose walls fail and fill with plaque.
When Willis fed guinea pigs a diet stripped of vitamin C, with no added cholesterol, they grew plaques identical to human atherosclerosis, and when he gave the vitamin back, the lesions cleared. He titled his 1957 report, plainly, The Reversibility of Atherosclerosis. Then he went further. In 1954, photographing the same arteries months apart, he put patients on 500 mg of vitamin C three times a day. Of 6 untreated controls, none improved and 3 worsened. Of 10 treated, 6 had plaques visibly shrink, and in one a leg artery opened enough to raise calculated flow nearly eightfold. Only the young, soft plaques reversed, not the calcified ones. It was, as far as the record shows, the first time anyone had imaged the regression of atherosclerosis in a living person.
Willis also performed autopsies and found human arterial tissue focally depleted of vitamin C exactly at the points of mechanical stress, a local, hidden scurvy in people with no outward sign of disease. You did not need to be a sailor dying of scurvy to be scorbutic where it counted. You only needed to be deficient in the wall of one coronary artery.
His hypothesis tied it together. Where the wall runs short of vitamin C, the ground substance that cements its lining turns leaky, and blood cholesterol drifts in and binds where it never should. In the guinea pigs the deficient diet alone produced the lesions while their blood cholesterol barely moved, and a radiolabeled tracer proved the cholesterol in the plaque came from the bloodstream, not the artery. The plaque was not the artery making fat. It was the blood seeping into a wound vitamin C should have kept closed.
And then medicine moved on. The cholesterol hypothesis was ascendant, the rabbit model was convenient, and Willis and his guinea pigs were filed away and forgotten for a generation.
The Surrogate
“Lipoprotein(a) is a surrogate for ascorbate.”
Rath and Pauling, PNAS, 1990
By the late 1980s two new pieces had appeared. First, arterial disease was understood to begin not as a slow greasing of a clean pipe but as a lesion, a crack in the wall. Second, a sticky, dangerous variant of LDL called lipoprotein(a), written Lp(a), had been characterized, and was found to anchor to tissue through lysine binding sites.
Pauling and Matthias Rath connected the two findings to Willis’s old work, and in 1990 published their hypothesis in the Proceedings of the National Academy of Sciences. Lp(a), they proposed, is a surrogate for ascorbate. The reasoning ran like this. Lp(a) appears in significant amounts almost only in the same short list of species that lost the ability to make their own vitamin C, the primates and the guinea pig. That coincidence, they argued, is not an accident.
When vitamin C is plentiful, the body builds strong collagen, because vitamin C is the cofactor that turns lysine and proline into the hydroxylysine and hydroxyproline that lock collagen fibers together. When it runs short, the wall weakens and the body reaches for an emergency patch. That patch is Lp(a), sticky enough to plaster over a leaking vessel. For an animal facing a brief winter of scarcity, a brilliant stopgap. In a human mildly deficient every day for 50 years, the stopgap never stops. The patch builds on the patch, and that is what we call plaque.
“The body was not failing. It was improvising. Lp(a) was the splint it reached for when the vitamin that builds arteries ran out.”
In a second 1990 PNAS paper, Rath and Pauling supplied the direct evidence. Guinea pigs deprived of vitamin C accumulated Lp(a) inside their arterial lesions, while guinea pigs given an adequate intake of vitamin C developed neither the lesions nor the Lp(a) deposits. The surrogate appeared exactly when the vitamin disappeared, and vanished when the vitamin returned.
The Cure
“Ascorbate prevents the development of atherosclerotic lesions in this animal model.””
Rath and Pauling, hypoascorbemic guinea pig study, 1990
Here is where Pauling, the chemist, made the leap no cardiologist had. If Lp(a) anchors to the wall through lysine binding sites, the move is obvious. Flood the blood with free lysine, and the loose molecules occupy those sites, blocking new Lp(a) and prying loose what has stuck. Rath added proline on the same logic.
So the proposed cure had three parts, each cheap and non toxic. Vitamin C in gram doses to rebuild the collagen so the wall stops needing patches. Lysine, 2 to 6 grams a day, as an Lp(a) binding inhibitor. And proline alongside it. Case reports of angina easing on the combination accumulated from 1991 onward, though they remained anecdotes. Pauling, never modest, believed the regimen could bring cardiovascular disease under nearly complete control.
There was a second half to the theory. Pauling and Rath argued the weakened wall is patched not by Lp(a) alone but by Lp(a) and fibrin together. In their 1991 paper they named the primary lesion the deposition of lipoprotein(a) and fibrinogen in the vascular wall. Lp(a) docks onto that fibrin through the same lysine binding sites, because its apo(a) protein is a near twin of plasminogen, the body’s own clot dissolving enzyme. By occupying plasminogen’s parking spots, Lp(a) keeps the patch from ever being cleared. Clot and cholesterol are stitched together at the lysine site.

Food versus the bottle: the doses against what a plate can deliver
Pauling therapy doses; food values from USDA and legume data
The table raises an obvious question. Why 6 grams of vitamin C when the blood saturates near 400 mg? Pauling’s answer was that the saturation figure comes from healthy young men, while a stressed, diseased body burns through it far faster, and because the vitamin clears within hours, only repeated grams keep the wall saturated. The honest rebuttal is that the kidney excretes most of an oral megadose, and no trial has shown the high dose beats a modest one for the heart.
This is why lysine is the hinge of the whole cure. Free lysine floods those binding sites, blocks Lp(a) from gluing itself to fibrin and the wall, and can strip loose what has already stuck. Vitamin C does two jobs at once. It rebuilds the collagen so the wall no longer needs a patch, and it converts the lysine residues in the wall into hydroxylysine, lowering their affinity so Lp(a) cannot grip. Lysine and proline release the patch, and vitamin C makes sure the wall stops asking for one. Rath and Pauling were confident enough in the chemistry to patent the approach in the early 1990s.
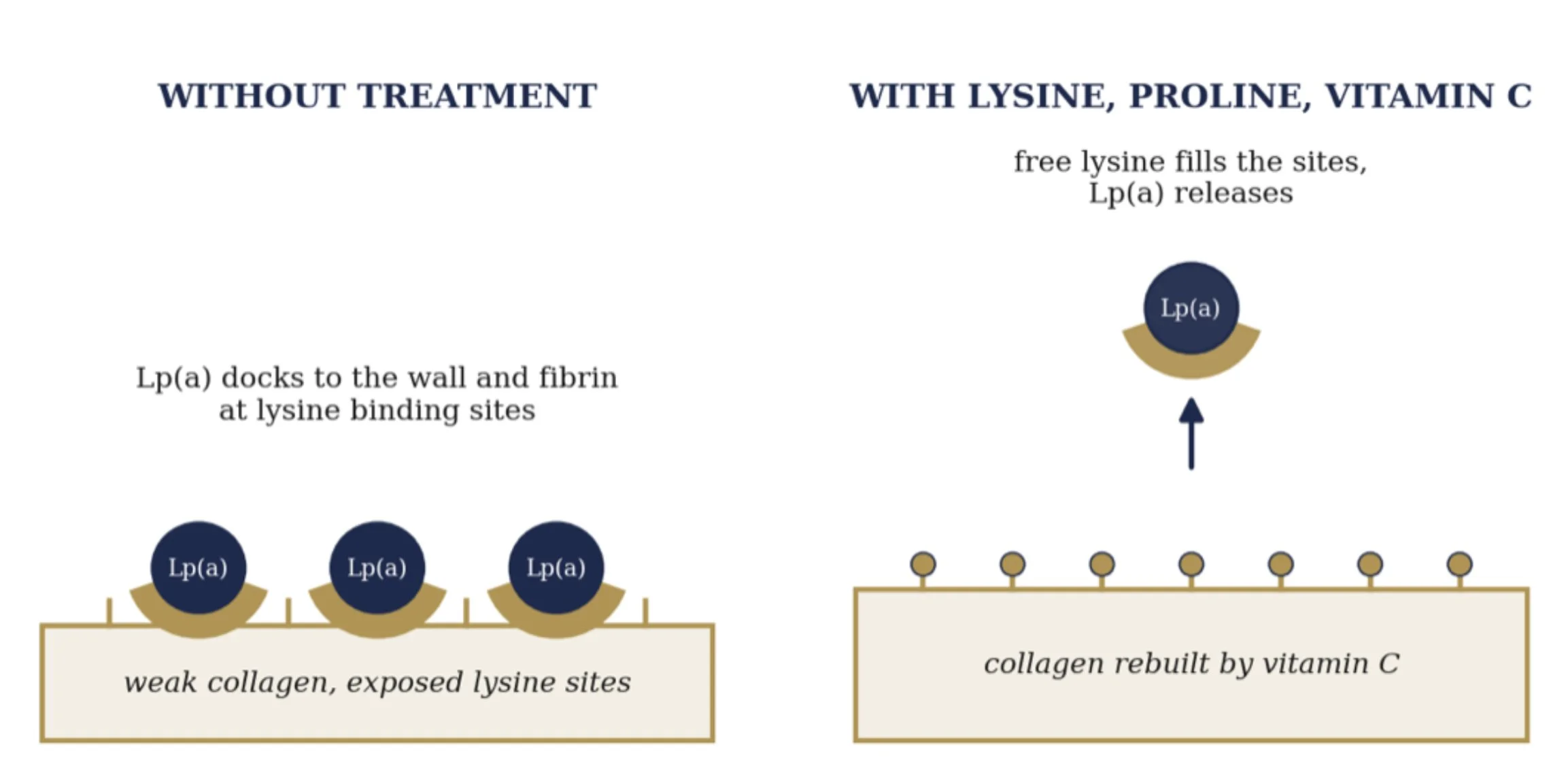
How vitamin C, lysine, and proline release Lp(a) from the wall
Mechanism after Rath and Pauling, 1991 and 1992
It was, on its face, the most consequential medical claim of the century. A two time Nobel laureate had named the killer’s weapon, explained the mechanism, and handed the world a cure made of vitamins and amino acids. And organized medicine declined to pick it up.
How LP(a) Kills
“Lp(a) is about six times more atherogenic than LDL.”
JACC, 2024
To see why this molecule is so lethal, look at how it is built. Lp(a) is an ordinary LDL particle with a second protein, apolipoprotein(a), bolted on. That protein is the whole problem, because it is a near copy of plasminogen, the enzyme the body uses to dissolve clots, sharing as much as 70 percent of its structure. From that one quirk come three ways to cause a heart attack.
The first weapon is plaque. Like LDL, Lp(a) slips into the artery wall and is trapped and oxidized there, only worse. On an equal particle basis it is about six times more atherogenic than ordinary LDL. This is the slow lane, the patient construction of the blockage that Willis and Pauling described.
The second weapon is fire. Lp(a) is the body’s main carrier of oxidized phospholipids, the inflammatory cargo that switches on macrophages and floods the wall with cytokines, leaving a plaque unstable, thin capped, and prone to crack. A particle that merely narrowed arteries would be survivable. One that also inflames them is what makes them rupture.
The third weapon is unique to Lp(a), and it is the one that kills. Because apo(a) impersonates plasminogen, it blocks the conversion of plasminogen to plasmin and raises the body’s clot stabilizing signals. So at the very moment an inflamed plaque cracks, the body cannot dissolve the clot forming over it. The artery seals shut. That sealed artery is the heart attack.
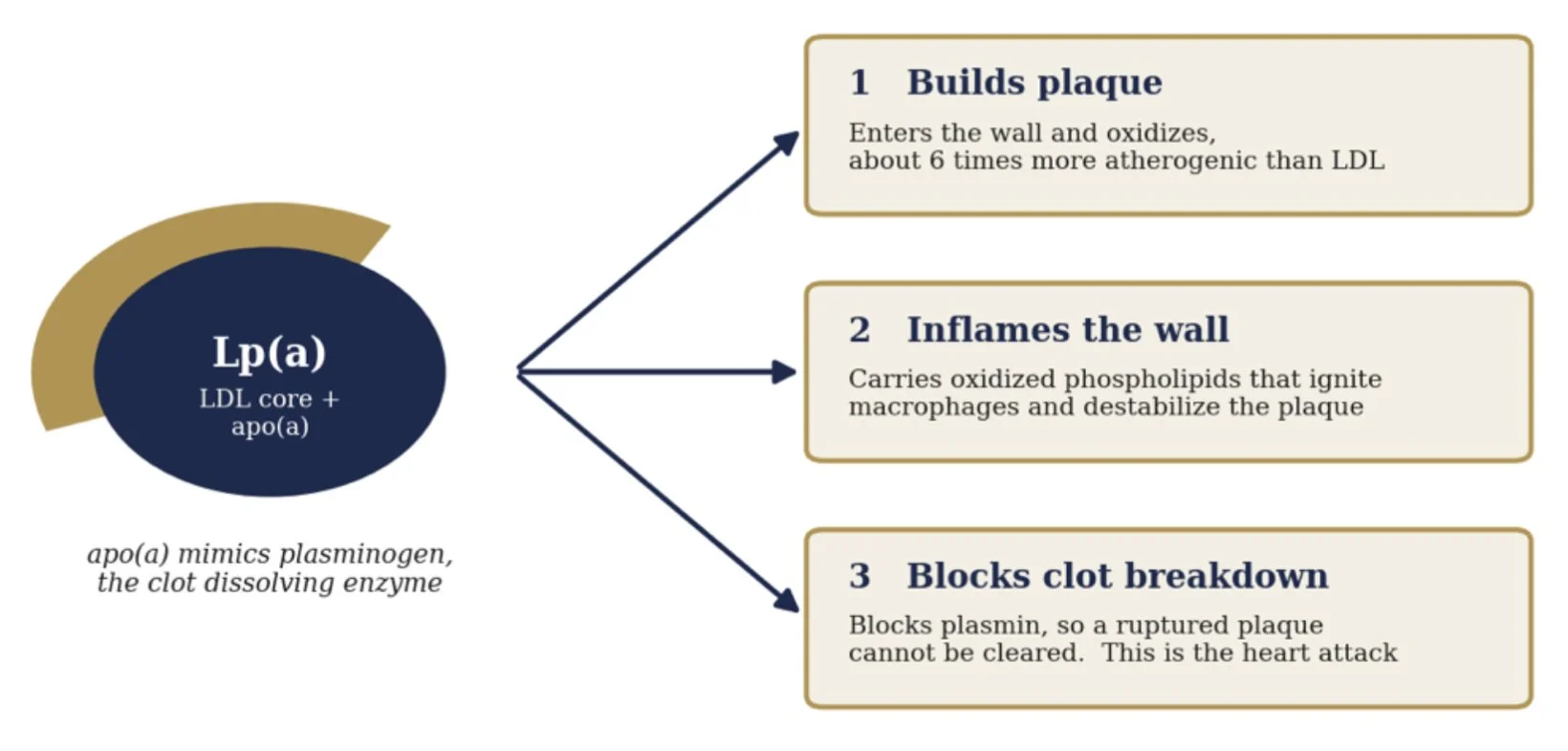
One particle, three ways to cause a heart attack
Mechanism after Boffa and Koschinsky, Nature Reviews Cardiology, 2019
The Genetic Verdict
“A genetically determined, causal, and prevalent risk factor.”
American Heart Association scientific statement, 2022
Here is the fact that turns Lp(a) into a moral one. You do not earn your Lp(a) level through how you live. You inherit it. Between 70 and over 90 percent of a person’s Lp(a) level is fixed by a single gene, the LPA gene, set at conception and holding nearly constant for the rest of life. Diet barely moves it. Exercise barely moves it. Statins do not move it.
That fixity let scientists prove, in a way they almost never can, that Lp(a) does not merely travel with heart disease but causes it, through Mendelian randomization. Because genes are dealt at random at conception and cannot be confounded by how you live, a variant that raises Lp(a) is a lifelong trial run by nature. If those dealt the high Lp(a) genes suffer more heart attacks, the molecule is guilty.
In 2009 the case was closed. The Emerging Risk Factors Collaboration pooled 36 studies and confirmed the association across a smooth gradient. The same year a Copenhagen group showed that genetically elevated Lp(a) raised the risk of myocardial infarction, and a team in the New England Journal found two LPA variants, rs10455872 and rs3798220, that raised coronary risk by roughly 50 to 90 percent per copy. The detail that sealed it: once they adjusted for Lp(a) level, the variants lost all predictive power. The genes act through the molecule and nothing else.
The relationship is a straight dose response. A doubling of Lp(a) raises heart attack risk by roughly 15 percent, and people above 100 mg/dL run more than double the risk of those near zero. The American Heart Association now calls Lp(a) a genetically determined, causal, and prevalent risk factor that stays dangerous even after LDL is driven down. Pauling pointed at this molecule in 1990. The genetics convicted it 20 years later.
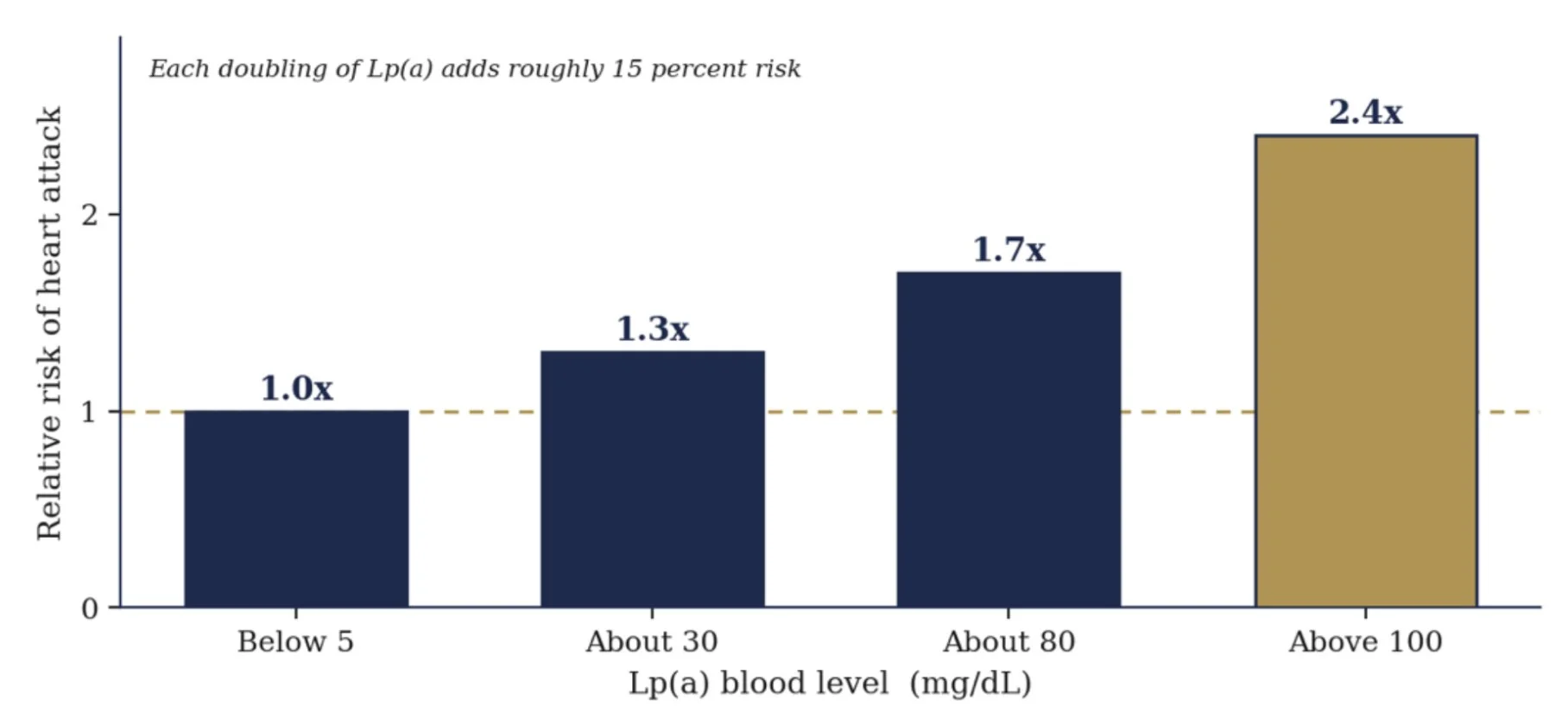
Heart attack risk climbs with Lp(a) level
Approximate, after Copenhagen population studies, Kamstrup 2009
Why Medicine Looked Away
It is worth being honest about why, because the reasons are not all foolish.
First, reputation. Through the 1970s and 1980s Pauling had championed high dose vitamin C for the common cold and then for cancer. Two Mayo Clinic trials, in the New England Journal of Medicine in 1979 and 1985, found it did nothing for advanced cancer. Pauling protested they had used the wrong route, oral instead of iv, but the community had seen enough, and his heart hypothesis inherited the stigma before it was ever tested.
Second, the evidence and the venue. The unified theory papers appeared largely in the Journal of Orthomolecular Medicine, outside mainstream cardiology. The human evidence was pilot data, case reports, and Pauling’s own testimony, never a large randomized trial. By the rules of evidence based medicine, and they are good rules, mechanism plus anecdote is a hypothesis, not a proof.
Third, the reigning paradigm. Medicine had committed its intellect and soon its pharmacy to LDL cholesterol and the statins. A theory insisting that the true driver was a vitamin deficiency and an obscure lipoprotein cut directly against a consensus that would become one of the largest enterprises in the history of medicine.
Fourth, money on the other side too. Rath built a supplement business around the protocol. That commercial entanglement, fair or not, gave academics an easy reason to keep the theory at arm’s length.
Fifth, timing. The tools to measure and to lower Lp(a) at scale, and to run an outcome trial around it, simply did not exist yet. Pauling died in 1994 with the heart hypothesis unproven and very nearly buried.
The Quiet Vindication
“An independent and causal risk factor for cardiovascular disease.”
National Lipid Association, on Lp(a), 2024
Then the molecule Pauling had blamed came back. In the last decade, cardiology has arrived, by its own road and its own data, at three conclusions that Pauling stated first.
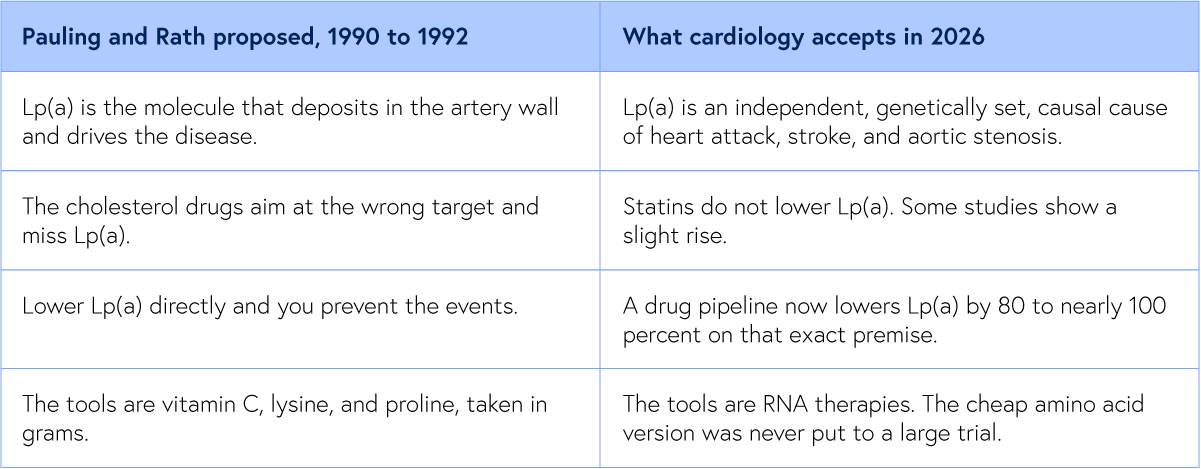
Lp(a) is now recognized as one of the strongest independent, genetically determined, causal risk factors for heart attack, stroke, and aortic valve disease, the culprit Pauling named 30 years before the field agreed. And the statins modern cardiology was built on do not lower it, blind to the very molecule Pauling fingered.
Most striking of all, the entire frontier of cardiovascular drug development is now aimed at doing exactly what Pauling proposed, lowering Lp(a) directly. A wave of new therapies, pelacarsen, olpasiran, lepodisiran, zerlasiran, and the oral muvalaplin, lowers Lp(a) by 80 to nearly 100 percent on precisely Pauling’s premise, that knocking down Lp(a) prevents heart attacks and strokes. The field simply chose engineered RNA over lysine and vitamin C.
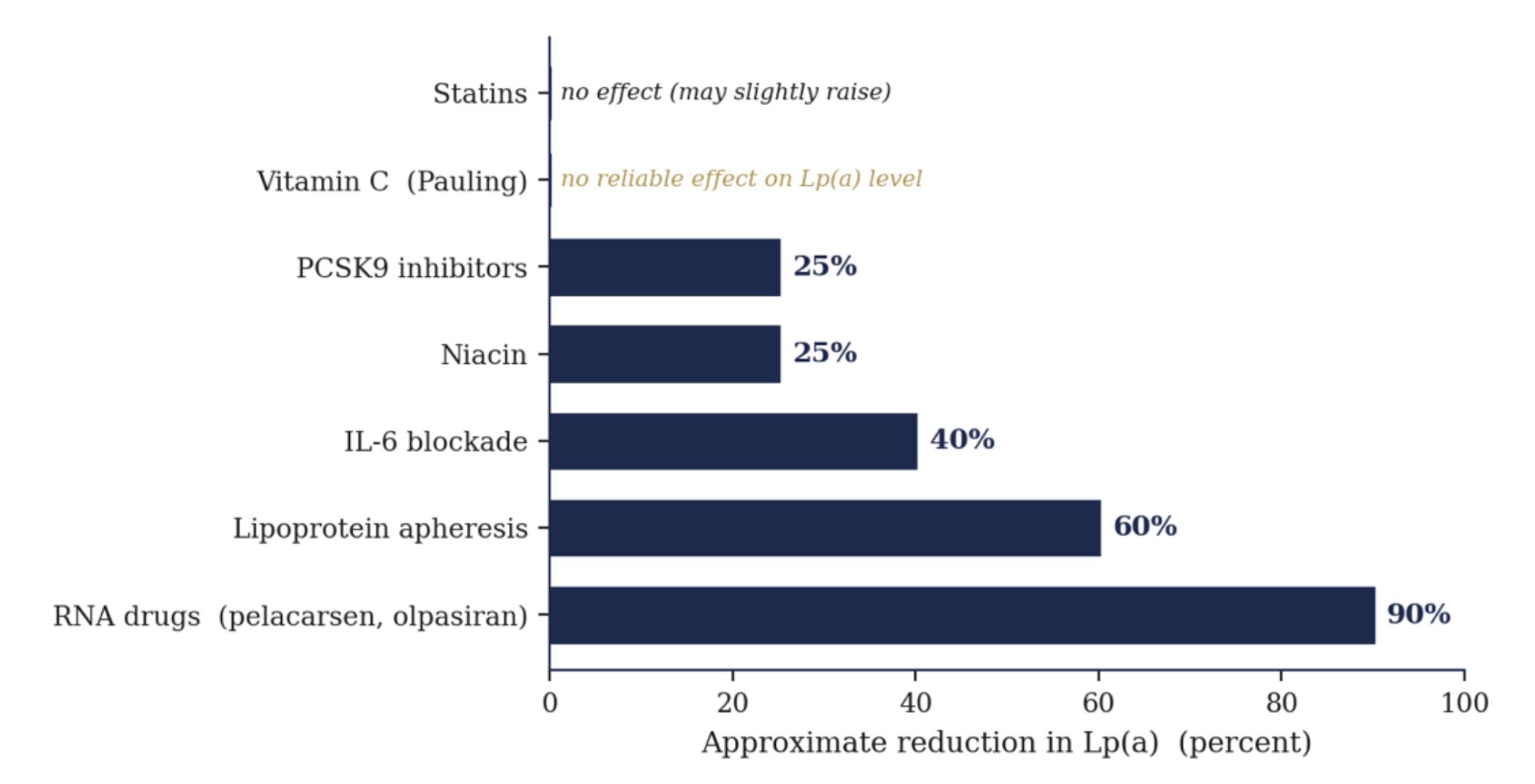
What actually lowers Lp(a), from statins to the new drugs
Approximate reductions. Neither statins nor vitamin C lower the level
Lower the Risk
“Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease.”
Boffa and Koschinsky, Nature Reviews Cardiology, 2019
If the number is written in your genes, is there anything to be done short of the new drugs? Yes, and it comes from separating two things that are easily confused. The level of Lp(a) is mostly fixed. The damage it does is not.
Inflammation is the most actionable lever. The LPA gene is not deaf to the body around it. It carries response elements for interleukin 6, the master inflammatory signal, so chronic inflammation actively drives Lp(a) production. In patients given an interleukin 6 blocking drug, Lp(a) fell by as much as 30 to 50 percent, proof that the level is not wholly beyond reach. And since much of Lp(a)’s harm is delivered through inflammation in the first place, calming the fire blunts both the number and the damage at once.
Oxidation is the next lever, with a caveat. The cargo Lp(a) carries is oxidized phospholipids, so the danger is partly oxidative state, not only quantity. The honest catch is that megadose antioxidant pills have repeatedly failed to lower events in trials, so the credible move is reducing the body’s whole oxidative load through diet and living, not trusting one supplement to neutralize the payload.
Epigenetics is the frontier. The fact that inflammation can switch the LPA gene up and down is proof it is regulated, not frozen. Whether those marks can be deliberately shifted by how we eat and live is still an open question, but it is a real reason the inherited number is not a closed sentence.
This part of the story is mine to act on. I cannot rewrite the LPA gene I was given. Although it’s normal, I can neutralize any of it and possibly even hasten the reversal of the plaque, as it sounds theoretically possible, based on first principle and not much downside from my perspective except focus and time.
I can lower the inflammation that amplifies it, reduce the oxidation that arms it, and clear the fibrin it deposits, with an anti-inflammatory whole food diet, with vitamin C and lysine and proline at the binding sites, and nattokinase at the clot. Cut the cause. Allow the healing. Strengthen the wall. Track the number. The molecule is inherited. The outcome is negotiated.
What It Means
It would be too easy, and untrue, to declare Pauling simply right. The honest verdict is more interesting and more humbling. He was almost certainly right about the target, and he was years ahead of everyone on the mechanism. Lp(a) is real, it is causal, and lowering it is now the great hope of preventive cardiology.
But two questions remain open. The first belongs to all of cardiology. No outcome trial has yet confirmed that lowering Lp(a) reduces events, though the answer is arriving now, with the Lp(a) HORIZON trial of pelacarsen reading out around 2026 and OCEAN(a) by 2027. The second belongs to Pauling alone. No one with the means ever ran a large outcome trial of his cheap amino acid version. Parts of it have not held up in isolation, since vitamin C alone does not appear to lower Lp(a), but the regimen as a whole was never tested. The ingredients are cheap and long off patent, so no sponsor funded the trial.
That gap, between a nearly free intervention and the absence of a trial to confirm or refute it, is the lesson of this story. The artery does not know whether its Lp(a) was lowered by a vitamin or a molecule that took a billion dollars to design. Pauling understood the chemistry. What he could not supply was the will of a system to test a cure no one could sell.
For my own protocol the conclusion is simple. I do not take vitamin C, lysine, and proline as a finished cure. I take them as a wager with good odds and almost no downside, and I give the two time Nobel laureate his due, late, but at last.
A Request
Each Friday, I upload a new Youtube video. Please like, comment and subscribe so I can help many others in your network and beyond, it’s my mission to help people avoid the same fate as Rob, the same fate as I could have had. Heart attack, stroke or sudden death.
https://www.youtube.com/@DrKevinHam
My latest video is going viral :) My #1 meal to unclog arterial plaque. Thank you.
What to Start Now
Get the lipoprotein(a) test. Lp(a). Your doctor likely has not heard of it.
Reduce your risk by lowering your total LDL risk, lowering oxidation, glycation (cut out refined carbs and added sugars) and inflammation (hsCRP<1.0)
Your Question
Question worth exercising with
For yourself. For someone you love. Answer them in the quietness of your day.
When will you get the Lp(a) blood test if you haven’t already?
For Someone You Love
There is someone in your life running and falling. You thought of them. Send this to them. Your loved ones just need the information to act and a guide to help them.
Keep going. The race is long, the road is beautiful, and the body was built to heal.
Grace, strength and love to you.
MORE READINGS YOU’LL ENJOY
Health
Reversing My 77% Heart Plaques
Stats Say You Likely Have Heart Plaque
The Healing Power of Food: Nitric Oxide
Meaning
I pray you unlock your heart to reach the height of your full potential by discovering your calling.
Kevin Ham, MD
Appendix
The summary of the referred exercise studies:
Willis GC. An Experimental Study of the Intimal Ground Substance in Atherosclerosis. Can Med Assoc J. 1953;69:17 to 22.
Willis argued that plaque forms over arterial ground substance weakened by vitamin C depletion and clusters at points of mechanical stress, framing atherosclerosis as a repair response in a locally scorbutic wall rather than a passive spill of fat.
Willis GC, Light AW, Gow WS. Serial Arteriography in Atherosclerosis. Can Med Assoc J. 1954;71:562 to 568.
On vitamin C at 500 mg three times a day, 6 of 10 treated patients showed plaque regression on serial arteriography, while none of the 6 untreated controls improved and 3 worsened. In one case a femoral lumen widened enough to raise calculated flow nearly eightfold. The first serial imaging evidence that human atherosclerosis is reversible.
Willis GC, Fishman S. Ascorbic Acid Content of Human Arterial Tissue. Can Med Assoc J. 1955;72:500 to 503.
Measuring vitamin C in arterial tissue at autopsy, Willis found it locally depleted at the sites of mechanical stress where plaque forms, evidence of a focal scurvy in the artery wall even in people with no systemic signs of scurvy.
Willis GC. The Reversibility of Atherosclerosis. Can Med Assoc J. 1957;77:106 to 108.
In guinea pigs, which like humans cannot make vitamin C, withdrawal produced atherosclerosis and replacement reversed it completely. In 6 of 10 human patients tracked by arteriography, plaques regressed on supplemental vitamin C.
Rath M, Pauling L. Hypothesis: Lipoprotein(a) is a surrogate for ascorbate. Proc Natl Acad Sci USA. 1990;87:6204 to 6207.
Rath and Pauling proposed that Lp(a) acts as a stand in for vitamin C in the species that lost the ability to make ascorbate, depositing on weakened vessel walls as an emergency patch when collagen synthesis falters. Useful in scarcity, it becomes plaque under chronic deficiency.
Rath M, Pauling L. Immunological evidence for the accumulation of lipoprotein(a) in the atherosclerotic lesion of the hypoascorbemic guinea pig. Proc Natl Acad Sci USA. 1990;87:9388 to 9390.
Direct evidence for the hypothesis: vitamin C deprived guinea pigs accumulated Lp(a) inside their arterial lesions, while an adequate intake of vitamin C prevented both the lesions and the Lp(a) deposits. The authors argued for an analogous mechanism in humans.
Rath M, Pauling L. Solution to the Puzzle of Human Cardiovascular Disease: Its Primary Cause Is Ascorbate Deficiency Leading to the Deposition of Lipoprotein(a) and Fibrinogen/Fibrin in the Vascular Wall. J Orthomolecular Med. 1991;6:125 to 134.
The fuller theory, naming fibrinogen and fibrin as the co deposit alongside Lp(a) and proposing that lysine and synthetic lysine analogs, taken with vitamin C, can release Lp(a) from its bonds to the wall. The chemical basis of the lysine binding cure.
Rath M, Pauling L. A Unified Theory of Human Cardiovascular Disease. J Orthomolecular Med. 1992;7:5 to 15.
The capstone synthesis naming chronic ascorbate deficiency as the primary cause of cardiovascular disease, with Lp(a) and fibrin deposition as the downstream damage, and vitamin C, lysine, and proline as the proposed therapeutic counter.
Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412 to 423.
A pooled analysis of 36 prospective studies establishing that higher Lp(a) tracks with higher risk of heart attack and coronary death across a continuous gradient. The large scale observational foundation for everything that followed.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331 to 2339.
A Copenhagen population study showing that LPA gene variants which raise Lp(a) also raise the risk of myocardial infarction. A landmark Mendelian randomization result moving Lp(a) from associated to causal.
Clarke R, et al. (PROCARDIS). Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518 to 2528.
Identified two LPA variants (rs10455872 and rs3798220) that raise Lp(a) and coronary risk, each copy adding roughly 50 to 90 percent. Crucially, the variants lost their association once Lp(a) level was accounted for, proving the molecule itself is the cause.
Burgess S, et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a) Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018;3:619 to 627.
Used genetics to estimate how much Lp(a) must be lowered to matter clinically, on the order of 100 mg/dL, directly shaping the design and dosing of the new Lp(a) lowering drug trials.
Reyes-Soffer G, et al. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease. AHA Scientific Statement. Arterioscler Thromb Vasc Biol. 2022;42:e48 to e60.
The American Heart Association consensus statement, declaring Lp(a) genetically determined (70 to over 90 percent), causal, and common, and noting it remains a risk even after LDL is effectively lowered.
Boffa MB, Koschinsky ML. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat Rev Cardiol. 2019;16:305 to 318.
Proposes that the oxidized phospholipids carried by Lp(a) tie its three harms together, the atherogenic, inflammatory, and thrombogenic arms, into a single mechanism. The intellectual frame for treating the damage rather than only the level.
Schulte DM, et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein(a) expression and lipoprotein(a) synthesis in humans. J Lipid Res. 2015;56:1034 to 1042.
Showed that interleukin 6 drives LPA gene expression and that blocking it lowers Lp(a), while a different anti inflammatory drug did not. Evidence that inflammation actively regulates the Lp(a) level, not only its harm.
Moertel CG, et al. High dose vitamin C versus placebo in the treatment of patients with advanced cancer. N Engl J Med. 1985;312:137 to 141.
A double blind trial that found no benefit of high dose oral vitamin C in advanced cancer. Though about cancer, not the heart, its decisive negative result hardened mainstream skepticism toward Pauling and cast a long shadow over the heart hypothesis that followed.
Nordestgaard BG, Langsted A. Lipoprotein(a) and cardiovascular disease. Lancet. 2024;404:1255 to 1264.
A modern review establishing Lp(a) as an independent, largely genetic, causal risk factor for atherosclerotic disease and aortic stenosis, and noting that conventional lipid lowering drugs, including statins, do not meaningfully lower it.
Lp(a) HORIZON trial of pelacarsen, NCT04023552. OCEAN(a) trial of olpasiran, NCT05581303.
The pivotal phase 3 outcome trials of the new Lp(a) lowering drugs, the first designed to test whether large reductions in Lp(a) actually prevent heart attacks and strokes. Results are expected between 2026 and 2027 and will judge the premise Pauling stated first.
Subscribe to my Compounding Wisdom newsletter and start transforming your life. ham.com
Subscribe to my YouTube channel @DrKevinHamfor videos on how I reversed my clogged arteries in 3 months, the top foods that clear your arteries, and the first principles of health that can save your life. Like, share and subscribe — it could save the life of someone you love.